Molecular Dynamics¶
The MolecularDynamics propagator performs a molecular dynamics simulation,
using some specific algorithms. These algorithms are sumarized on the
Algorithms used in MolecularDynamics propagator figure. These algorithms are presented in this documentation
section.
The MolecularDynamics type have the following constructors:
-
MolecularDynamics(timestep)¶ Creates a molecular dynamics propagator using a Velocity-Verlet integrator with the specified timestep. Without any thermostat or barostat, this performs a NVE integration of the system.
-
MolecularDynamics(::Integrator) Creates a
MolecularDynamicspropagator with the specified integrator.
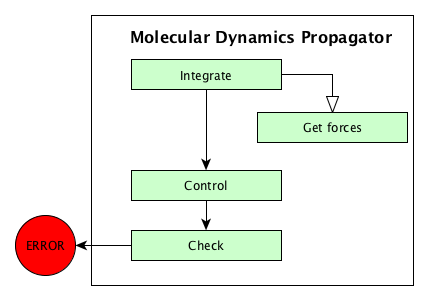
Algorithms used in MolecularDynamics propagator
Time integration¶
An integrator is an algorithm responsible for updating the positions and the
velocities of the current frame of the universe. It is represented by a subtype of the Integrator type. You can
set the integrator to use with your simulation using the set_integrator!()
function.
Verlet integrators¶
Verlet integrators are based on Taylor expensions of Newton’s second law. They provide a simple way to integrate the movement, and conserve the energy if a sufficently small timestep is used. Assuming the absence of barostat and thermostat, they provide a NVE integration.
-
type
Verlet¶ The Verlet algorithm is described here for example. The main constructor for this integrator is
Verlet(timestep), wheretimestepis the timestep in femtosecond.
-
type
VelocityVerlet¶ The velocity-Verlet algorithm is descibed extensively in the literature, for example in this webpages. The main constructor for this integrator is
VelocityVerlet(timestep), wheretimestepis the integration timestep in femtosecond. This is the default integration algorithm in Jumos.
Force computation¶
The NaiveForces algorithm computes the forces by iterating over all the pairs of
atoms, and calling the appropriate interaction potential. This algorithm is the
default in Jumos.
Controlling the simulation¶
While running a simulation, we often want to have control over some simulation
parameters: the temperature, the pressure, etc. This is the goal of the control
algorithms, all subtypes of the Control type.
Controlling the temperature: Thermostats¶
Various algorithms are available to control the temperature of a simulation and perform NVT simulations. The following thermostating algorithms are currently implemented:
-
type
VelocityRescaleThermostat¶ The velocity rescale algorithm controls the temperature by rescaling all the velocities when the temperature differs exceedingly from the desired temperature.
The constructor takes two parameters: the desired temperature and a tolerance interval. If the absolute difference between the current temperature and the desired temperature is larger than the tolerance, this algorithm rescales the velocities.
sim = Simulation(MolecularDynamics(2.0)) # This sets the temperature to 300K, with a tolerance of 50K thermostat = VelocityRescaleThermostat(300, 50) push!(sim, thermostat)
-
type
BerendsenThermostat¶ The berendsen thermostat sets the simulation temperature by exponentially relaxing to a desired temperature. A more complete description of this algorithm can be found in the original article [1].
The constructor takes as parameters the desired temperature, and the coupling parameter, expressed in simulation timestep units. A coupling parameter of 100, will give a coupling time of \(150\ fs\) if the simulation timestep is \(1.5\ fs\), and a coupling time of \(200\ fs\) if the timestep is \(2.0\ fs\).
-
BerendsenThermostat(T[, coupling = 100]) Creates a Berendsen thermostat at the temperature
Twith a coupling parameter ofcoupling.sim = Simulation(MolecularDynamic(2.0)) # This sets the temperature to 300K thermostat = BerendsenThermostat(300) push!(sim, thermostat)
| [1] | H.J.C. Berendsen, et al. J. Chem Phys 81, 3684 (1984); doi: 10.1063/1.448118 |
Controlling the pressure: Barostats¶
Barostats provides a way to implement NPT integration. None of them is implemented in Jumos for now.
Checking the simulation consistency¶
Molecular dynamic is usually a garbage in, garbage out set of algorithms. The numeric and physical issues are not caught by the algorithm themselves, and the physical (and chemical) consistency of the simulation should be checked often.
In Jumos, this is achieved by the Check algorithms, which are presented in
this section. Checking algorithms can be added to a simulation by using the
push! function.
Existing checks¶
-
type
GlobalVelocityIsNull¶ This algorithm checks if the global velocity (the total moment of inertia) is null for the current simulation. The absolute tolerance is \(10^{-5}\ A/fs\).
-
type
TotalForceIsNull¶ This algorithm checks if the sum of the forces is null for the current simulation. The absolute tolerance is \(10^{-5}\ uma \cdot A/fs^2\).
-
type
AllPositionsAreDefined¶ This algorithm checks is all the positions and all the velocities are defined numbers, i.e. if all the values are not infinity or the
NaN(not a number) values.This algorithm is used by default by all the molecular dynamic simulation.
Default algorithms¶
Default algorithms for molecular dynamic are presented in the following table:
| Simulation step | Default algorithms |
|---|---|
| Integration | Velocity-Verlet |
| Forces computation | Naive computation |
| Control | None |
| Check | None |
Functions for algorithms selection¶
The following functions are used to to select specific algorithms for the simulation. They allow to add and change all the algorithms, even in the middle of a simulation run.
-
set_integrator!(sim, integrator)¶ Sets the simulation integrator to
integrator.Usage example:
sim = Simulation(MolecularDynamic(0.5)) run!(sim, 300) # Run with a 0.5 fs timestep set_integrator!(sim, Verlet(1.5)) run!(sim, 3000) # Run with a 1.5 fs timestep